
Tutorial
This tutorial introduces how to use this web server to predict and analyze possible type-III effectors. The web server allows anonymous use. If you want to keep your query sequences confidential and manage your jobs, please register and login using username.
This website can be divided into three parts.
The first part is BEAN 2.0. BEAN 2.0 uses features learned from known type-III effectors to predict query proteins as effectors or non-effectors.
The second part is designed for the further analysis of proteins, including four components. i) By searching a protein sequence against the Pfam library of HMMs, you can determine which domains it carries. ii) TargetP predicts the subcellular location of eukaryotic proteins. iii) Plant-mPLoc and Hum-mPloc predict subcellular localizations of plant and non-plant proteins including those with multiple sites, respectively. v)IUpred is used to predict the likelihood of disorder for each residues.
At last, we present a specialized database of annotated type-III effectors.

In the navigation bar, there are also some other conclusive information. For example, the nets of the domains and effectors are showed in Domain-net and Effector-net, respectively. Users can check the details of the networks just by spinning the wheels of the mouse. Users are also allowed to target at only one network by put the mouse on the node of this network.
In addition, you may click "Contribution" to make suggestions and provide effectors. We are very grateful for your suggestions and sharing. If you have some other questions, please don't hesitate to contact me.
FASTA format
BEAN 2.0 uses sequences in FASTA format to predict possible type-III effectors. FASTA is one of the most common formats that are used to represent DNA or protein sequences in modern biology. A FASTA sequence should be presented as:
>IPAD_SHIFL MNITTLTNSISTSSFSPNNTNGSSTETVNSDIKTTTSSHPVSSLTMLNDTLHNIRTTNQA LKKELSQKTLTKTSLEEIALHSSQISMDVNKSAQLLDILSRNEYPINKDARELLHSAPKE AELDGDQMISHRELWAKIANSINDINEQYLKVYEHAVSSYTQMYQDFSAVLSSLAGWISP GGNDGNSVKLQVNSLKKALEELKEKYKDKPLYPANNTVSQEQANKWLTELGGTIGKVSQK NGGYVVSINMTPIDNMLKSLDNLGGNGEVVLDNAKYQAWNAGFSAEDETMKNNLQTLVQK YSNANSIFDNLVKVLSSTISSCTDTDKLFLHF
The first line starts with a ">", and follow with the sequence name. The other lines are the protein sequence represented in single letter form. If you have more than one sequence, you just need to write each of them one by one in the same way (Please don't leave any blank lines between them!).
BEAN 2.0
Using webserver
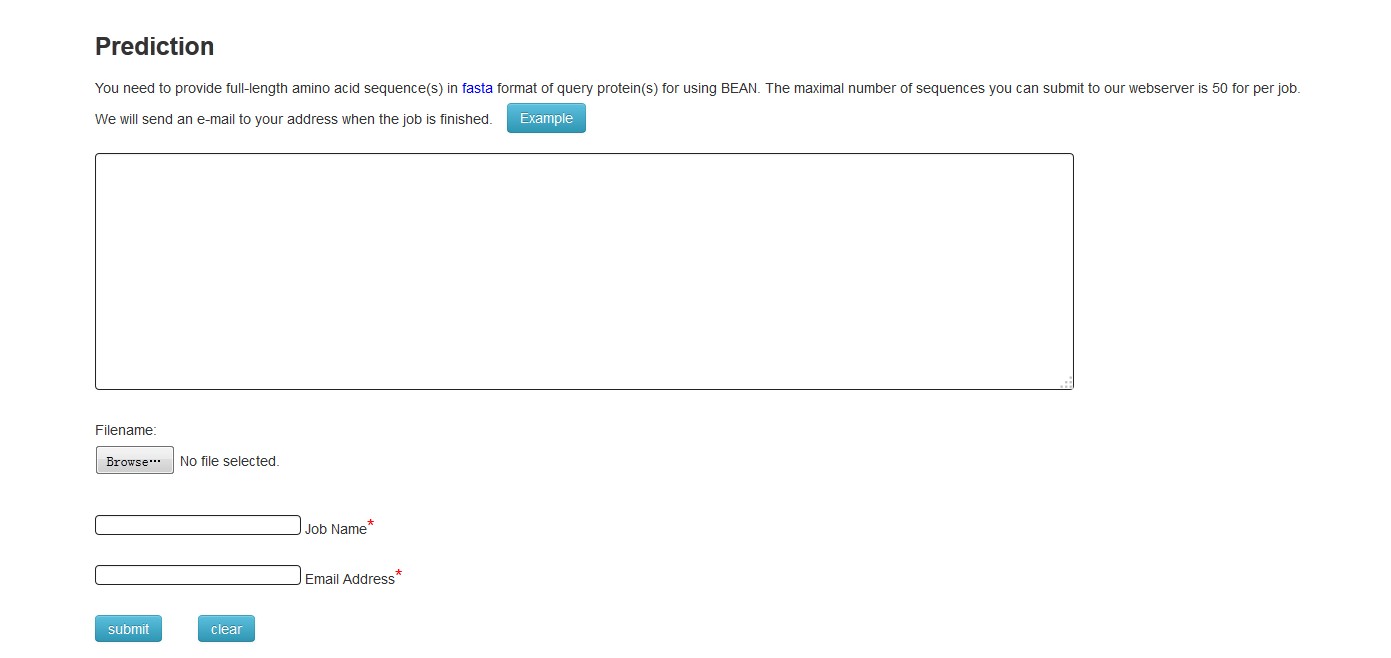
Click "start new job" to predict candidate proteins. You need to input your protein sequences in FASTA format into the textfield. The maximal number of sequences you can submit to our webserver is 50 for per job. Then please input your email address and submit. Each sequence may take 2 minutes or more. When your job is finished, you will receive an email. BEAN 2.0 will return the result reporting whether the query proteins are effectors or not in the webpage that is included in the email. Then you can download the results or further analyze the corresponding domains, subcellular locations and disorder residues of these proteins.
You can also check submitted jobs. You can login the system and manage your jobs if you have a account.
Running BEAN 2.0 on your local machine
Please refer to the installation guide wrapped in BEAN 2.0's software package or turn to download webpage.
Output
Algorithm
Click "wiki" to see the algorithm of BEAN 2.0.
Methods
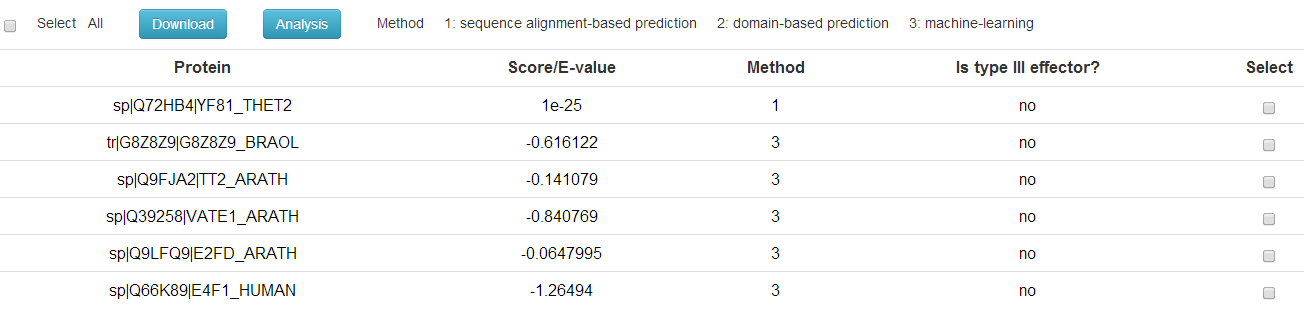
BEAN 2.0 consists of three components: sequence alignment-based predictor, domain-based predictor, and machine-learning predictor.
If the candidate protein is predicted by sequence alignment-based predictor(BLAST) or domain-based predictor(Pfam), e-value will be showed to
indicate the confidence level of the result. Otherwise, raw score will be used
to classify a unknown protein sequence as candidate type-III effectors or not.
1: sequence alignment-based prediction
2: domain-based prediction
3: machine-learning
Raw Score
BEAN* uses the raw score to classify a unknown protein sequence as candidate type-III effectors or not. The higher the score is, the more possible the protein is a type-III effector. BEAN 2.0 uses score=0.0 as a cutoff to make the decision, whereby all proteins assigned with scores equal or above 0.0 are considered as candidate TTEs.
E-value
BLAST and Pfam use E-value as a threshold to judge whether the candidate proteins can be predicted as effectors. The Expect value used as a convenient way to create a significance threshold for reporting results. The lower the E-value, the more significant the match is. The closer it is to zero, the results tend to be more reliable.
Analysis
Using webserver
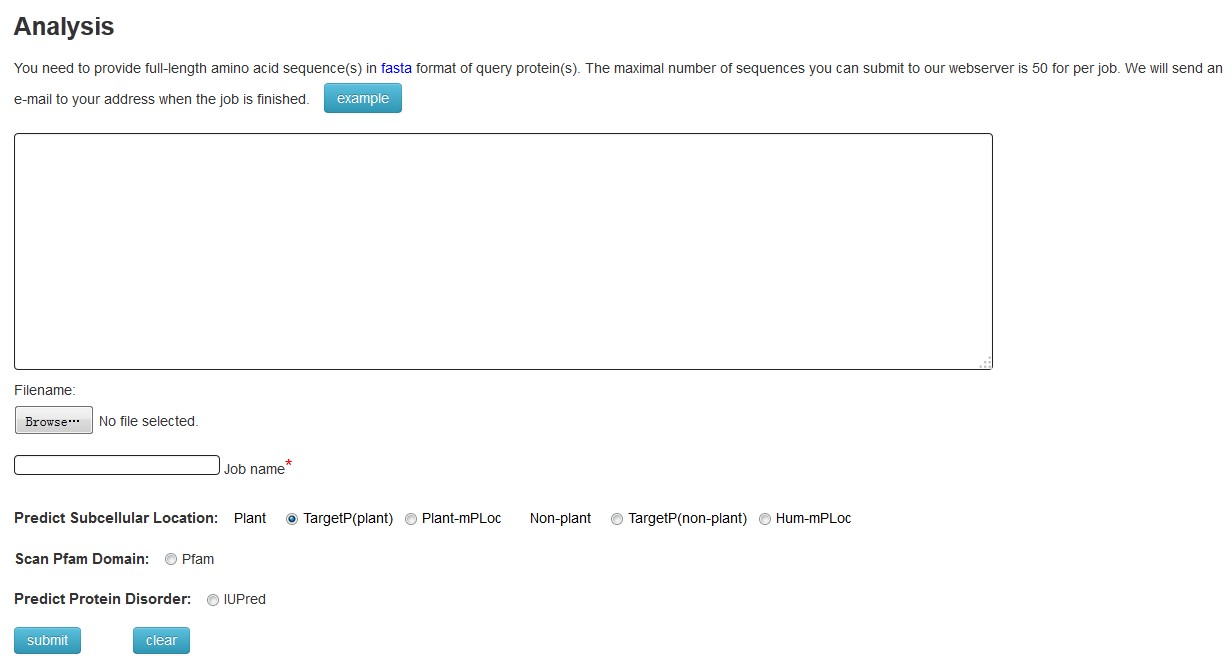
Click "analysis" to predict the domains, subcellular locations and disorder residues of query proteins. You need to input your protein sequences in FASTA format into the textfield, and give a name consisted with alphanumeric characters or underscores to demark you job. Then you also need to choose a type that you want to predict. After a few minutes ,you will get the result. You can also download your selected analysis results, too.
Tools
Pfam
The Pfam database is a large collection of protein domain families. Each family is represented by multiple sequence alignments and hidden Markov models (HMMs). By searching a protein sequence against the Pfam library of HMMs, you can determine which domains it carries.
TargetP
TargetP predicts the subcellular location of eukaryotic proteins. The location assignment is based on the predicted presence of any of the N-terminal presequences: chloroplast transit peptide (cTP), mitochondrial targeting peptide (mTP) or secretory pathway signal peptide (SP).
For the sequences predicted to contain an N-terminal presequence a potential cleavage site can also be predicted.
Plant-mPloc
Plant-mPLoc predict subcellular localizations of plant proteins including those with multiple sites.Hum-mPloc
Hum-mPloc predict subcellular localizations of non-plant proteins including those with multiple sites.IUPred
IUPred is a predictor of prediction of intrinsically unstructured proteins. Intrinsic disorder is particularly enriched in proteins implicated in some important functions.Output
Pfam
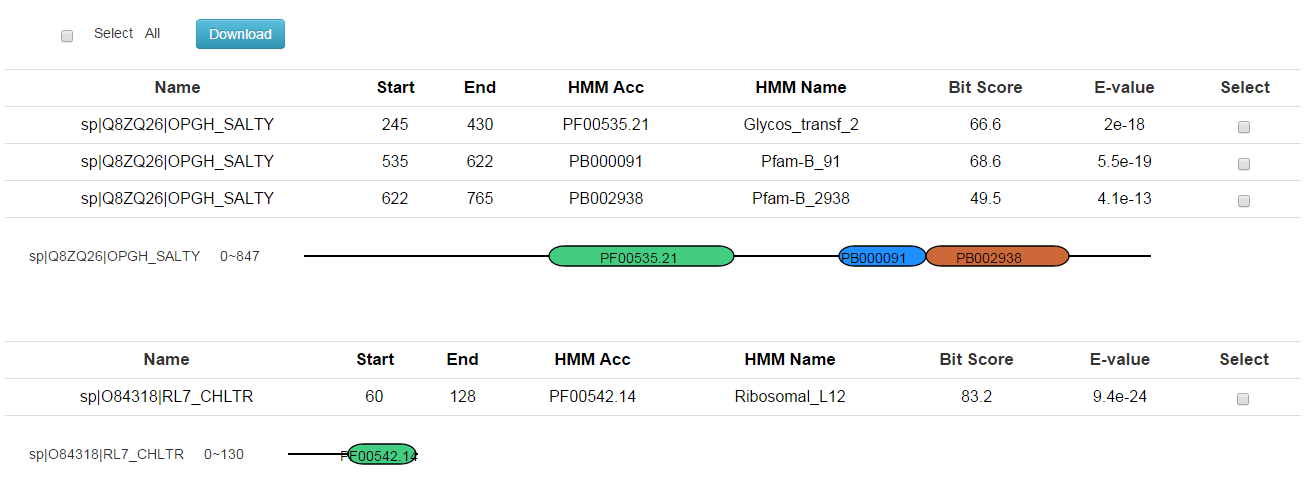
In addition to the results, you will get a graphical interface that displays the domains on the query proteins. For more information can be found at http://pfam.xfam.org/help.
TargetP
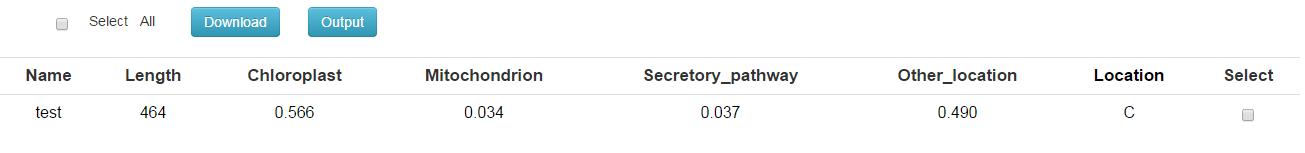
| Name | Sequence name truncated to 20 characters |
| Length | Sequence length |
| Chloroplast,Mitochondrion Secretory_pathway,Other_location | Final NN scores on which the final prediction is based. Note that the scores are not really probabilities, and they do not necessarily add to one. However, the location with the highest score is the most likely according to TargetP, and the relationship between the scores (the reliability class, see below) may be an indication of how certain the prediction is. |
| Location |
Prediction of localization, based on the scores above; the possible values are:
C Chloroplast, a chloroplast transit peptide; M Mitochondrion, a mitochondrial targeting peptide; S Secretory pathway, a signal peptide; _ Any other location; * "don't know"; indicates that cutoff restrictions were set (see instructions) and the winning network output score was below the requested cutoff for that category. |
For more information, please visit http://www.cbs.dtu.dk/services/TargetP/output.php.
Plant-mPloc
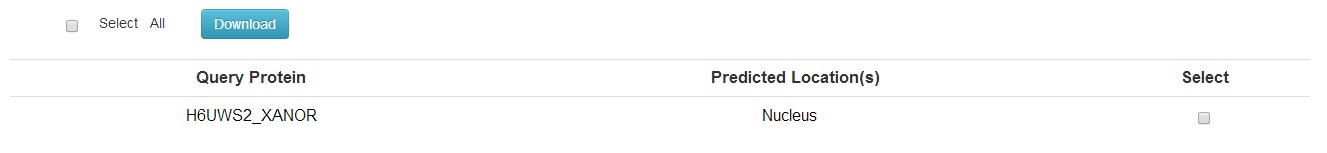
You will get a table contain the subcellular locations of the query proteins. Due to the instability of this predictor, the server will automatically turn to TargetP if there were no results after 100s.
Hum-mPloc
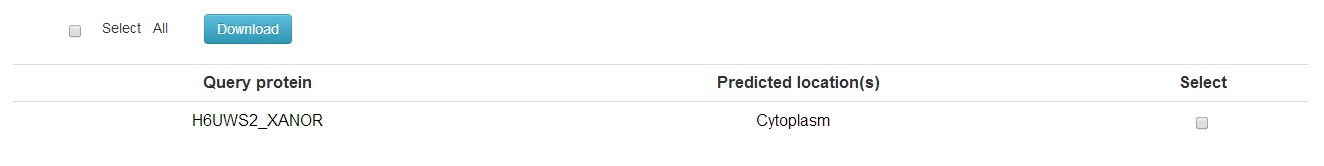
You will get a table contain the subcellular locations of the query proteins. Due to the instability of this predictor, the server will automatically turn to TargetP if there were no results after 100s.
IUPred
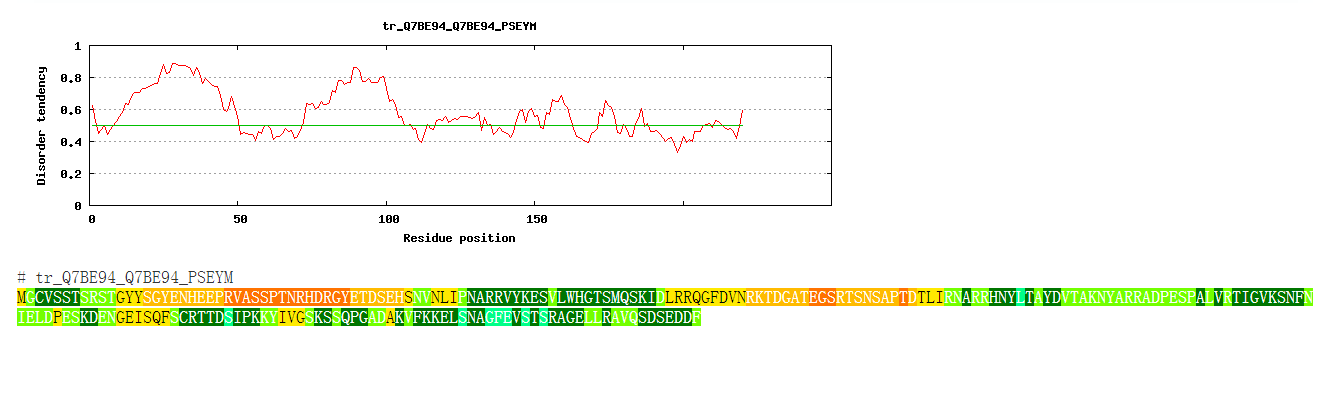
The output gives the likelihood of disorder for each residue, i.e. it is a value between 0 and 1, and higher values indicate higher probability of disorder. Residues with value above 0.5 can be regarded as disordered. The webserver showed the value use a rainbow bar. The color change from warm to cool with the increasing of the value.
Database
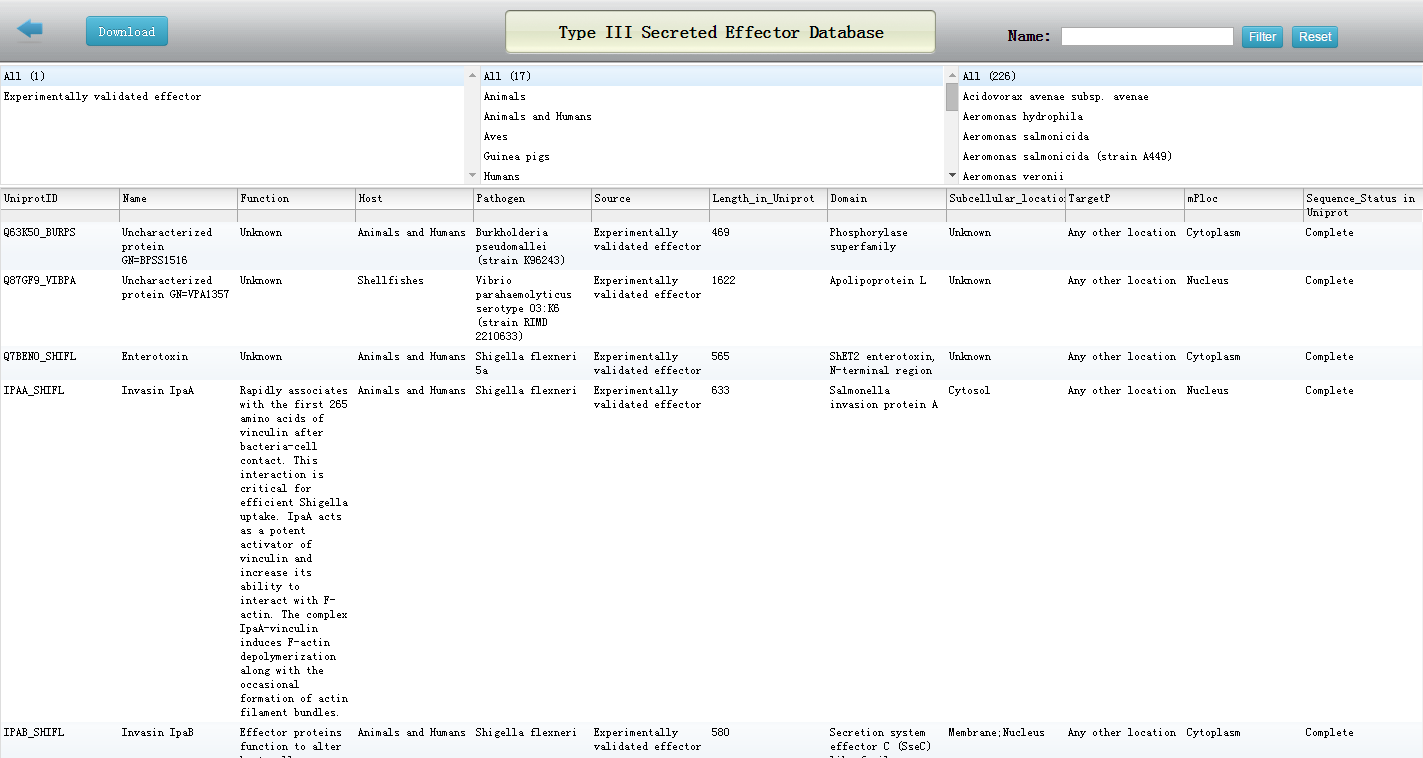
This database contains effector records. We collected these records mainly from Uniprot. Search results are presented in a tabular form, displaying effector name, source organism of the effector, sequence length, experimental status, domain(Pfam), subcellular locations in host. You can also download the database.
Exclusive domains in Type-III effectors have been summarized and displayed in Domain page.